
【NGS介紹系列-16S metagenome數據分析】
16S metagenome目前常用來進行環境微生物族群分析的方法,藉由微生物體內的高度保留16S rRNA基因的序列來進行研究,以便能夠以宏觀的角度進行探討,研究環境中菌相的變化。
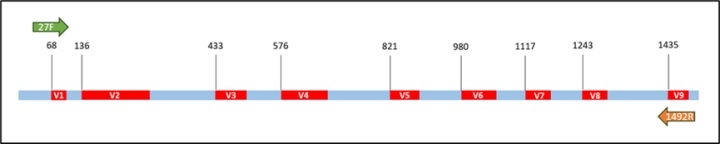
在16S rRNA基因上除了含有高度保留的區域之外,同時也存在著高度變異的區域 (如下圖),透過PCR的方式針對此區域進行放大 (常見區域為V3-V4區域),結合次世代定序的輔助進行定序。所獲得的數據依照原本PCR放大的片段大小進行處理,如果片段較大的話需要先進行Merge的步驟;如果片段較小的話可以直接使用單邊的序列進行後續分析。而依照不同的分析方法將相似的序列進行整合,獲得16S rDNA的Operational taxonomic unit (OTU)或是Amplicon sequence variants (ASV)等maker gene序列。此方式由於只著重在特定區域,此辨識的解析度會受到部分限制。近年來由於三代定序儀的現世,定序全長的16S rRNA基因日漸盛行,相較於前的特定區域的分析方式,此序列可以直接進行物種比對,鑑定的解析度也相較提高許多。
為了鑑定不同的微生物,16S的定序資料將會針對已知的微生物資料庫進行比對,透過比對到的資料庫訊息,進行界、門、綱、目、科、屬及種等不同層次的分類學分析。目前常使用的資料庫有SILVA、RDP及Greengenes等。如此將鑑別出來的物種進行比較,進而找出其組別之間存在的差異性,以了解其對環境的影響。
#詳盡的售後服務諮詢
#專屬的數據管理系統
#迅速的發送結果報告
#專業的生物資訊分析
#源資生技
#TriIBiotech
#16smetagenome
NGS & 第三代定序服務
Nanopore,Illumina長短序列機型齊全
高效率,高品質的好選擇
專業的樣品品管程序與文庫製備
專業的生物資訊分析與服務諮詢