
【NGS介紹系列-細菌WGS分析與應用】
發佈於 2022-9-16
在介紹過細菌WGS是如何進行的之後,今天來告訴大家實驗完成後得到的數據後續要如何處理呢?
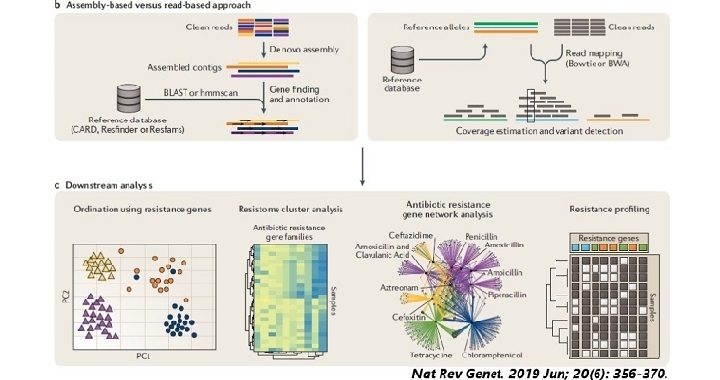
- 沒有參考序列(reference genome)時會使用De-novo assembly方法
- 有參考序列會使用Reference Mapping方法
1. De-novo assembly組裝
在沒有參考序列的情況下,僅使用測序的序列片段的資訊來組裝的方法。
利用將reads的末端序列相同的區域連結合併起來,形成contig
希望最後可以得到一條還原後的完整基因組序列(genome);
但是有些情況會使組裝遭遇困難,例如:定序錯誤、核酸多型性、定序偏差(sequencing bias)…….
這時候可以解決的方法為:
- 加深定序深度
- 組裝前先依據序列的品質分數(quality score)進行裁切(trim)
- 使用PCR free的方法製備樣品
2. Reference mapping
在有參考序列的情況下,將測序片段比對(mapping)至參考序列上,以取得分析結果。
Reference mapping的方式通常較為快速且對計算的要求較低,
對於參考序列的資料庫選擇非常重要,
分析結果的品質通常透過3個量化指標來評估:
- Mapping rate: Mapped reads/Total reads
- Genome coverage: Mapped genome/Total genome
- Depth: 平均深度及單鹼基的深度分布
近年來隨著NGS技術持續提升和生物資訊分析技術不斷進步,加上定序成本的降低,
細菌WGS在流行病學及公衛領域的使用逐漸廣泛,
如:抗藥性基因檢測、菌株基因分型、聚集事件調查、藥物疫苗開發…等
另外,在食品微生物的檢測方面,隨著大量微生物定序資料的快速累積,
能夠更有效率去追溯食品汙染源及傳播途徑,
甚至能藉此改善食品的製造或運輸程序以增進食品品質與安全,
NGS在疾病預防控制與民生食安上已成為不可或缺的工具了!
NGS & 第三代定序服務
Nanopore,Illumina長短序列機型齊全
高效率,高品質的好選擇
專業的樣品品管程序與文庫製備
專業的生物資訊分析與服務諮詢