

1984 年人類基因體計畫 (Human Genome Project, HGP) 以第一代定序方法(Sanger Sequencing),用了超過10年的時間且花費30 億美元完成整個人類基因體30億個鹼基對的定序。
隨著定序技術的演進與發展,2007年次世代定序 (Next Generation Sequencing, NGS) 的技術問世,大幅降低定序所需的成本和時間。次世代定序又稱為第二代定序或基因高通量分析(High-Throughput Analysis),此高效率的定序技術至今單次輸出量已可高達540-600 Gb (Illumina system),再結合上生物資訊的分析技術,次世代定序已被廣泛應用在各個不同的學術領域研究基因體、轉錄體及表觀基因體等。
第三代定序(Third Generation Sequencing),又可稱為「長讀取定序技術」(Long-Read Sequencing),目前平均的序列讀取長度可大於1 萬個鹼基(10 kb),能直接定序原始DNA 樣本,省去次世代定序技術需要的PCR增幅過程,可避開 PCR 擴增的錯誤率及偏好性問題,也能分析DNA 甲基化修飾。較廣為人知的技術有Pacific BioSciences (PacBio) 公司2010 年推出的「單分子即時定序」(Singlemolecule Real-Time Sequencing, SMRT),以及Oxford Nanopore Technologies (ONT) 公司於2012 年底推出的「奈米孔定序」(Nanopore-Based Platform)。第三代定序系統的解序片段長,除了上述的基因體、轉錄體、表觀基因體研究,於生物醫學和生物技術等領域具有更廣泛的應用。並且,第三代定序相對於傳統的一代、二代定序,在改善速度、降低成本和提高數據質量等方面也有顯著的進展,為研究人員的需求提供了更多的選擇。源資具備Nanopore平台的Flongle,MinION,GridION,以及2022年底最新推出的PromethION 2 Solo。
源資NGS & 第三代定序服務的優點 :
1. Nanopore,Illumina長短序列機型齊全
2. 高效率,高品質的好選擇
3. 專業的樣品品管程序與文庫製備
4. 專業的生物資訊分析與服務諮詢
- 想要進一步了解NGS & 第三代定序服務的相關問題,請點選 NGS常見問題。
- 若欲委託NGS & 第三代定序服務,請至本公司官網畫面 實驗室登入,登入後做進一步的訂購。
- 若需下載NGS & 第三代定序服務申請書相關檔案,請點選 檔案下載 !
Illumina

次世代測序 Next Generation Sequencing (NGS)
NGS (Next Generation Sequencing, NGS),又稱為次世代定序或基因高通量分析,以第一代定序方法為基礎而開發出的新技術。因第一代定序方法通量低、成本高與耗時長,對大規模之應用造成影響,因此有NGS技術的發展。NGS降低單一鹼基定序所需的成本,也讓當前的定序檢測不再受限於基因的大小或多寡。將想要分析序列片段化,接上定序標記序列完成序列文庫,即可轉入NGS定序儀,進行分子擴增,高通量定序技術可以搭配不同的文庫建構技術,應用各種基因體研究分析如Targeting gene sequencing、RNA sequencing、Metagenomics等多元應用。
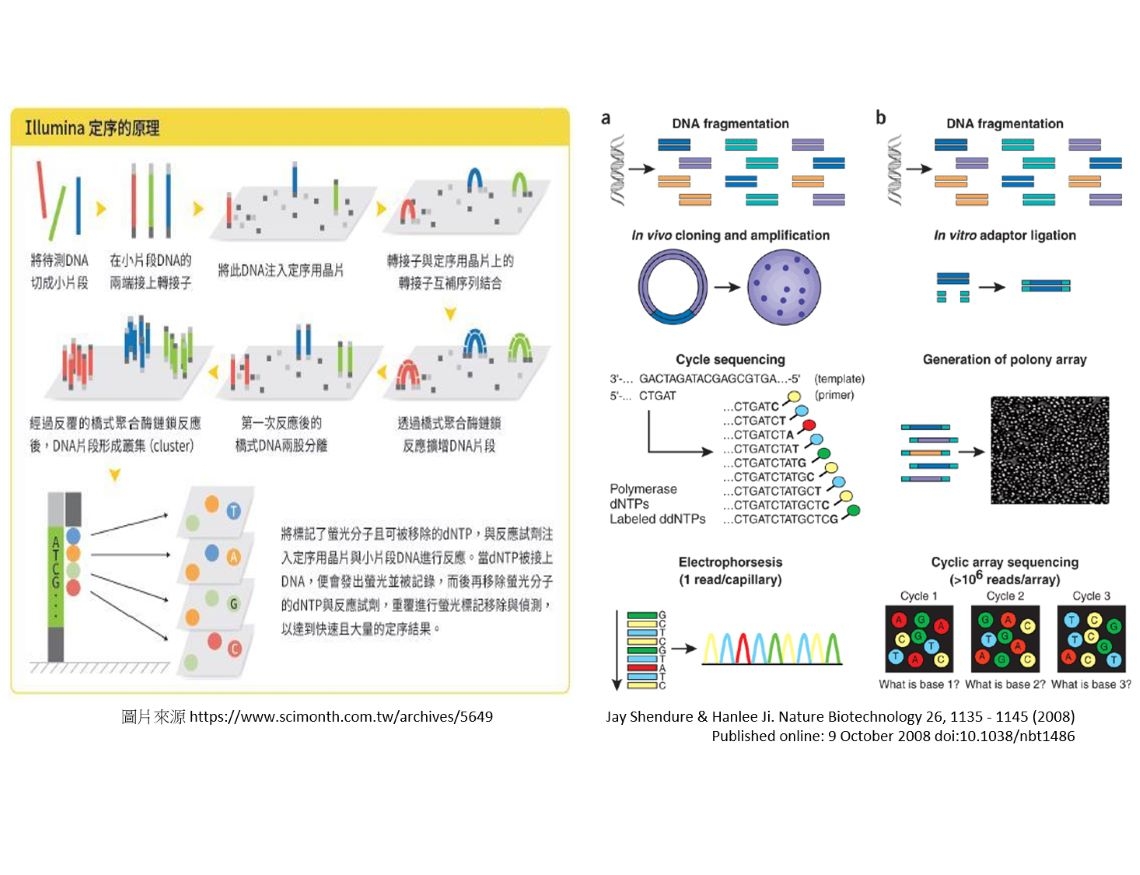
目前市面上,次世代測序以 Illumina推出的測序儀為主流,圖片為 Illumina測序原理簡介。
Nanopore

第三代測序 奈米孔定序 Nanopore
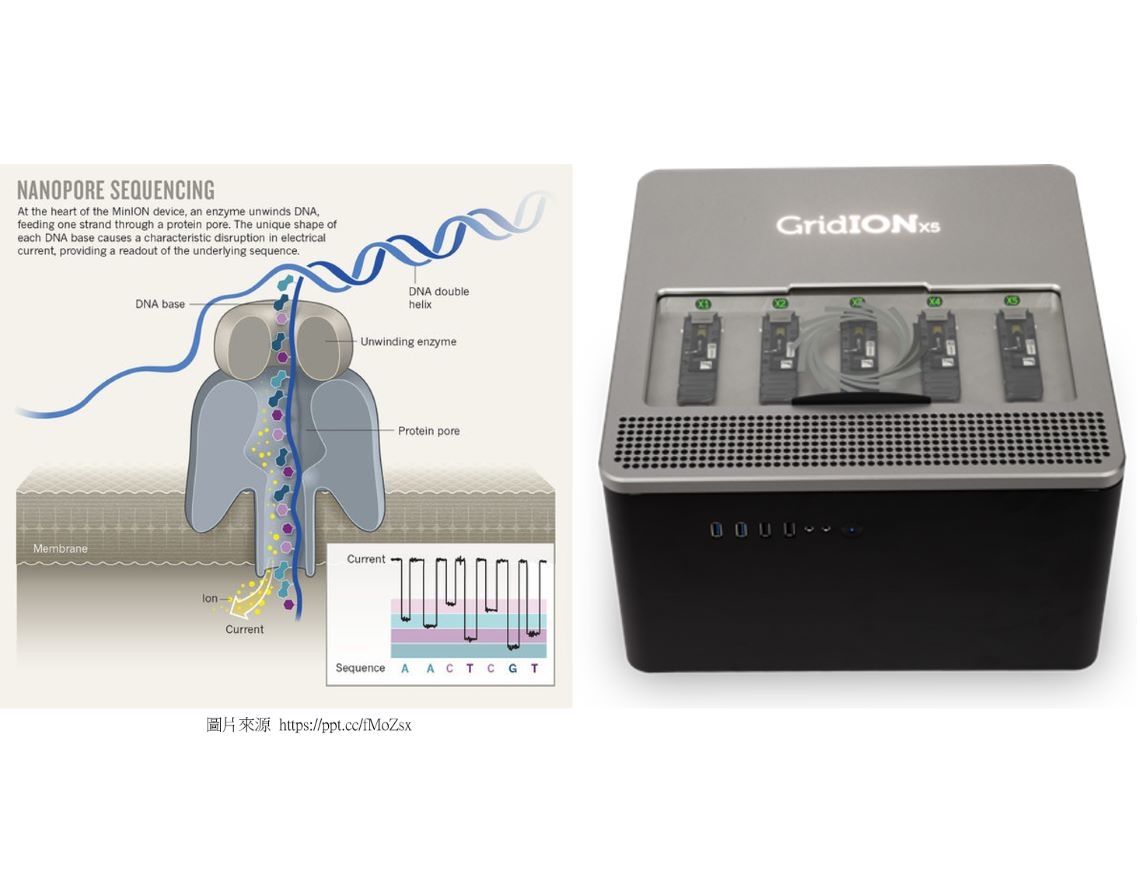
奈米孔定序(Nanopore sequencing),是一種針對核酸( RNA 與 DNA )進行定序的第三代定序技術,透過物理方法,利用特殊奈米孔洞內的共價鍵將分子結合在一起,當DNA或RNA鹼基通過奈米孔時會產生的電荷變化,進而短暫影響電流強度,即時監測電流強度變化來鑑別所通過的鹼基 。奈米孔定序技術不同於過去的基因定序技術,無須對樣品進行聚合酶鏈式反應或化學標記即可對一條DNA或RNA單個分子進行定序,能夠直接即時分析任意長度的DNA或RNA片段,且單條序列讀長可以超過1Mb,為提供基因體組裝帶來強而有力的助益。此外,奈米孔定序可以用相對低廉的成本對個體進行基因型分型,並具可攜帶性高、樣品處理的過程簡易快速與可以即時分析結果等優點。
目前奈米孔定序已被廣泛應用在各種領域,如人類基因體及甲基化定序、動植物基因體定序、病原菌基因體定序及快速檢測、環境或食品微生物監測等等其他相關應用。
Whole Genome Sequencing
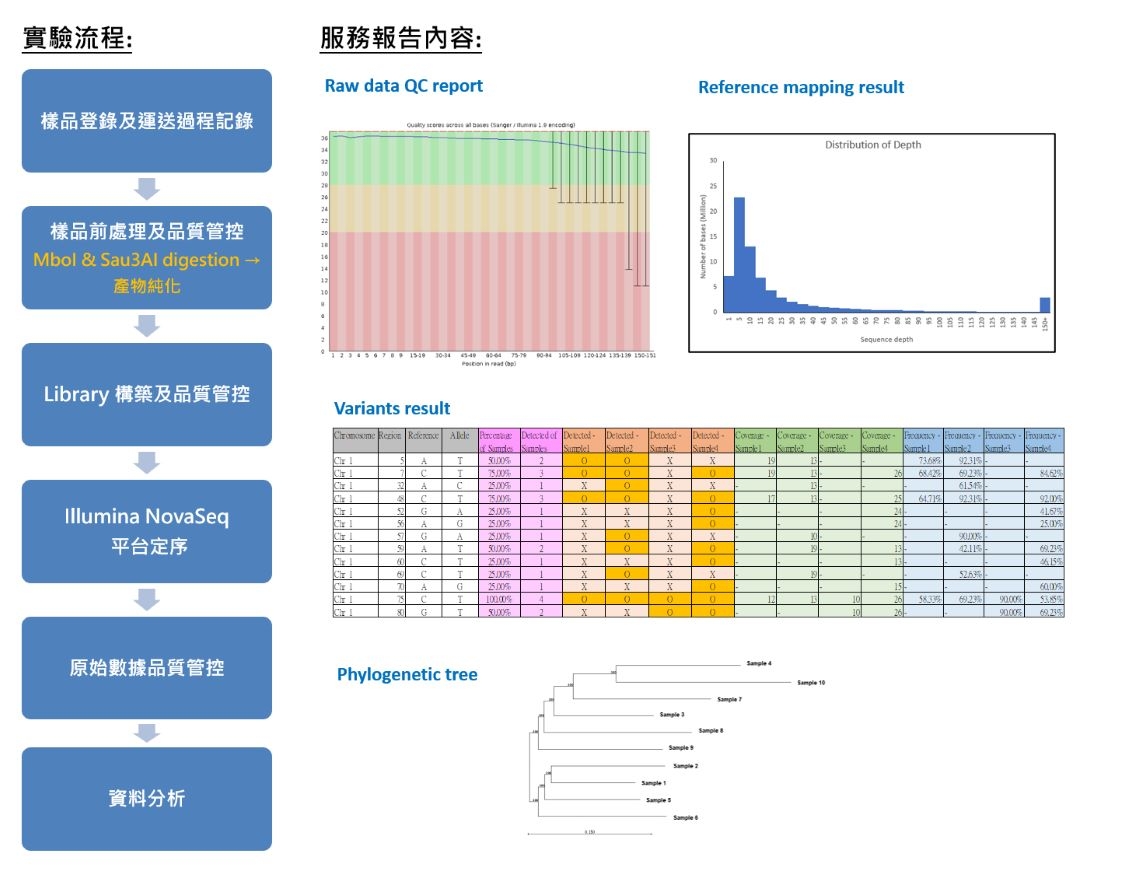
全基因組測序 Whole Genome Sequencing (Illumina)
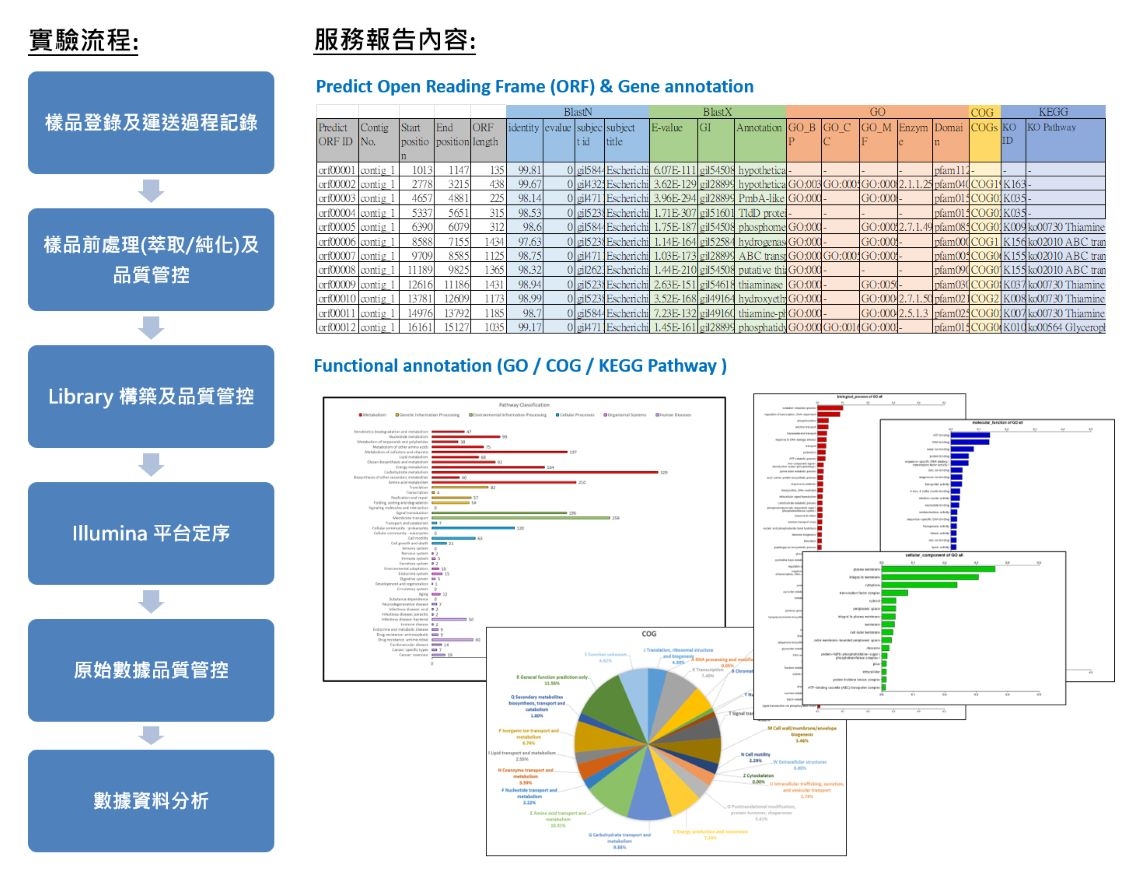
藉由次世代定序系統 Illumina 對DNA序列未知或已知的個體進行基因組測序,得知基因的序列,並獲得基因、遺傳變異、基因功能性以及藥物研究上的重要訊息,並可在個體或群體層次上進行差異性分析的方法探討疾病相關問題。
目前源資提供2種 Illumina上機平台,分別為MiSeq (PE300) 與NovaSeq (PE150)。進行全基因組測序時,會將樣本碎裂成適合測序的大小再進行Library構築及上機。報告內容包含樣本及數據的QC,序列的組裝與比對 (BlastN, BlastX),提供資料庫分析 (GO, COG, KEGG)及Genome map或者客製化分析服務。
樣品需求:
樣品純度:OD 260/280值應在1.7~1.9之間
樣品濃度:最低濃度不低 100 ng/µl
樣品總量:每個樣品總量至少1 µg(建議 5 µg)
樣品溶劑:可溶解在ddH2O或TE buffer(pH 8.0)中
全基因組測序 Whole Genome Sequencing (Nanopore)
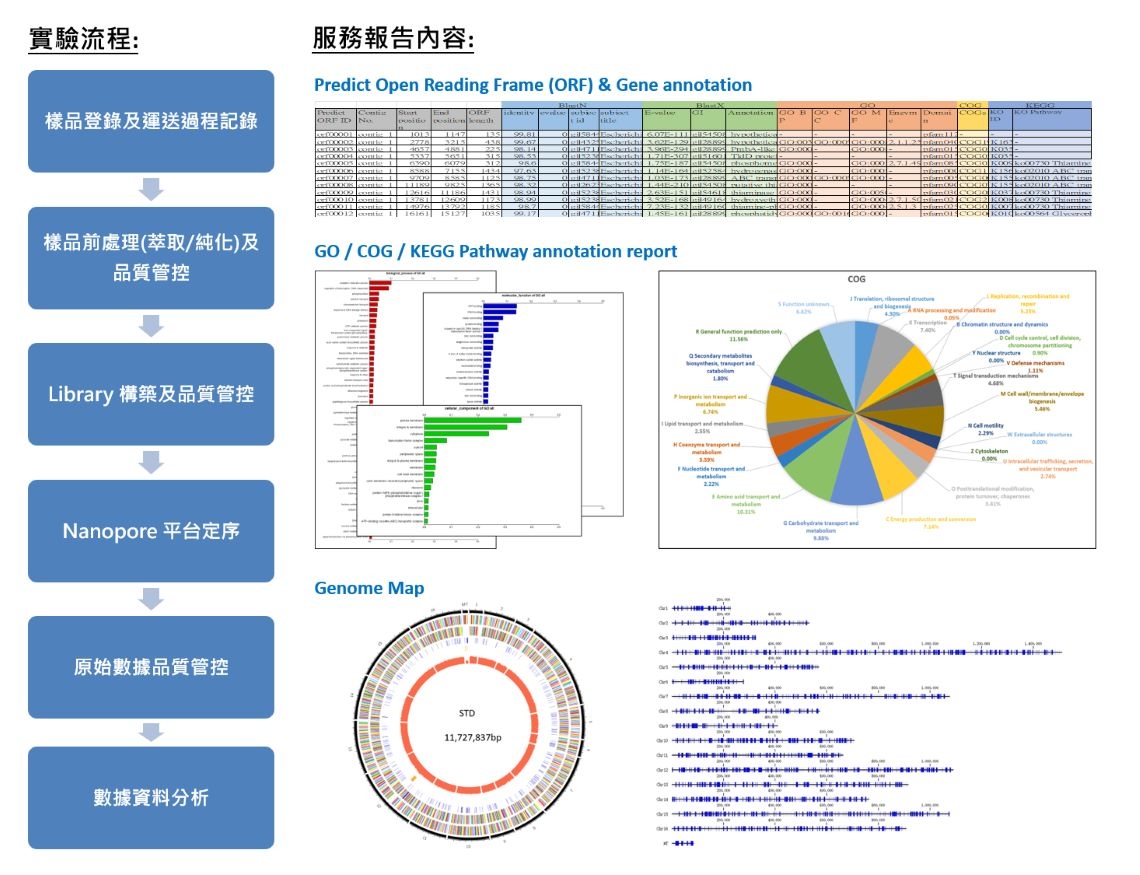
藉由三代定序平台 Nanopore 對 DNA 序列未知或已知的個體進行基因組測序。因Nanopore 測序數據長片段讀長的特性,在基因體組裝,結構變異區的判斷及連續重複序列的辨識上提供更好的效果,進而在基因體研究、生物醫學和生物技術等領域具有更廣泛的應用。
Nanopore上機可搭配不同規格的晶片做選擇。報告內容包含樣本及數據的QC,序列的組裝與比對 (BlastN, BlastX),提供資料庫分析 (GO, COG, KEGG)及Genome map或者客製化分析服務。
樣品需求:
樣品純度:OD 260/280值應在1.7~1.9之間
樣品濃度:最低濃度不低 100 ng/µl
樣品總量:每個樣品總量至少1 µg(建議 5 µg)
樣品溶劑:可溶解在ddH2O或TE buffer(pH 8.0)中
Whole Exome Sequencing
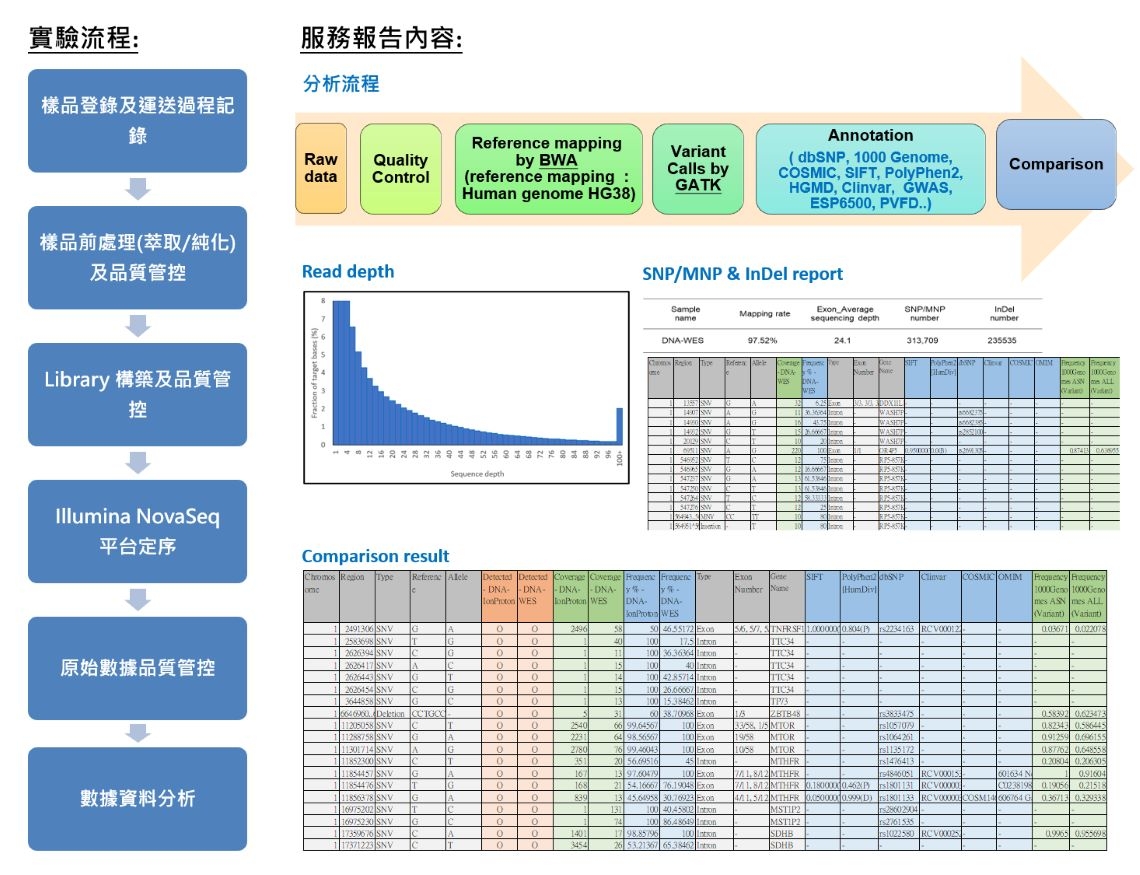
外顯子組測序 Whole Exome Sequencing (WES)
全外顯子測序(Whole Exome Sequencing, WES)是一種高通量的基因組技術,不同於全基因組測序(Whole Genome Sequencing, WGS),它主要集中於測序生物體的外顯子部分。外顯子是基因組中編碼蛋白質的區域,通常被認為是生物體功能最重要的基因區域。儘管整個人類基因組包含大約30億個鹼基對,但外顯子僅占總基因組的約1%,然而其中包含了大多數導致疾病的突變,也因此全外顯子測序對於識別蛋白質編碼基因的變異非常有價值,對於理解遺傳疾病和個體化醫療有重要貢獻。
全外顯子測序的主要特點包括:
1. 有針對性的測序: 與測序整個基因組不同,WES 選擇性地關注于基因的外顯子部分。外顯子是DNA的部分,會被轉錄成信使RNA(mRNA),最終用於合成蛋白質。
2. 經濟高效: 與全基因組測序(WGS)相比,WES 更為經濟,因為它只測序基因組的一小部分,但提供了有關蛋白質編碼區域的關鍵資訊,這在許多研究和臨床應用中非常有用。
3. 變異檢測: WES 主要用於檢測外顯子中的單核苷酸變異(SNVs)和小的插入或缺失(indels)。這些變異可以包括導致疾病的突變,也包括一些常見的遺傳多態性。
4. 臨床應用: WES 在鑒定罕見孟德爾遺傳病、癌症基因組學以及理解不同疾病的遺傳基礎方面具有重要的臨床應用。它有助於診斷和預測患者的疾病風險。
5. 癌症基因組學: WES 在癌症研究中常常用於識別腫瘤基因組中的體細胞突變,這有助於制定治療決策和開發靶向治療方法。
樣本需求:
樣品純度:OD 260/280值應在1.7~1.9之間;RNA、蛋白質應該去除乾淨
樣品總量:每個樣品總量建議1 µg以上(最少10 ng)
樣品溶劑:可溶解在ddH2O或TE buffer(pH 8.0)中。或是乾燥狀態下運送‧
16S/ITS Metagenomics
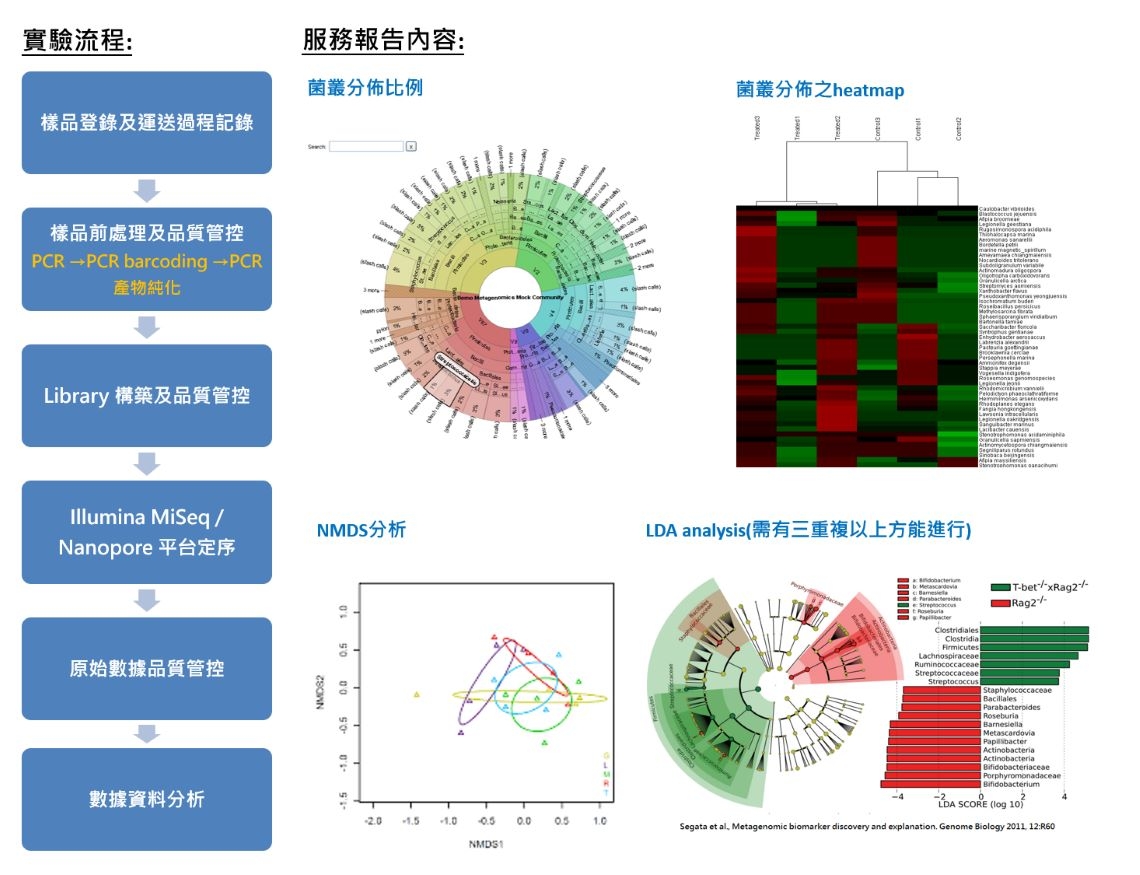
多源基因組學(宏基因組學) 16S/ITS Metagenomics
多源基因體學(Metagenomics),又稱為環境基因組學(Environmental Genomics),是研究一整個微生物群體的科學,從環境樣品萃取其中微生物的DNA加以定序後,然後進行物種鑑定、親緣、基因功能、表型關聯等分析比對,以了解環境所含微生物的多樣性。
16S rRNA基因存在於所有細菌的核糖體中,並且具有高度序列保守性和不同細菌的多樣性特徵,因此可用於分類和鑑定細菌。 16S metagenomics主要針對細菌的16S rRNA基因序列,經由PCR方式進行 rRNA片段放大,建構paired-end library ,以Illumina MiSeq平台進行解序,將所得序列進行分析,根據其序列變異和豐富度,得到各檢體所含的菌種種類及菌叢分佈比例。
樣品需求:
樣品純度:OD 260/280值應在1.7~1.9之間
樣品總量:建議每個樣品總量不少於 50 ng
樣品溶劑:可溶解在ddH2O或TE buffer(pH 8.0)中
Whole Genome Metagenomics
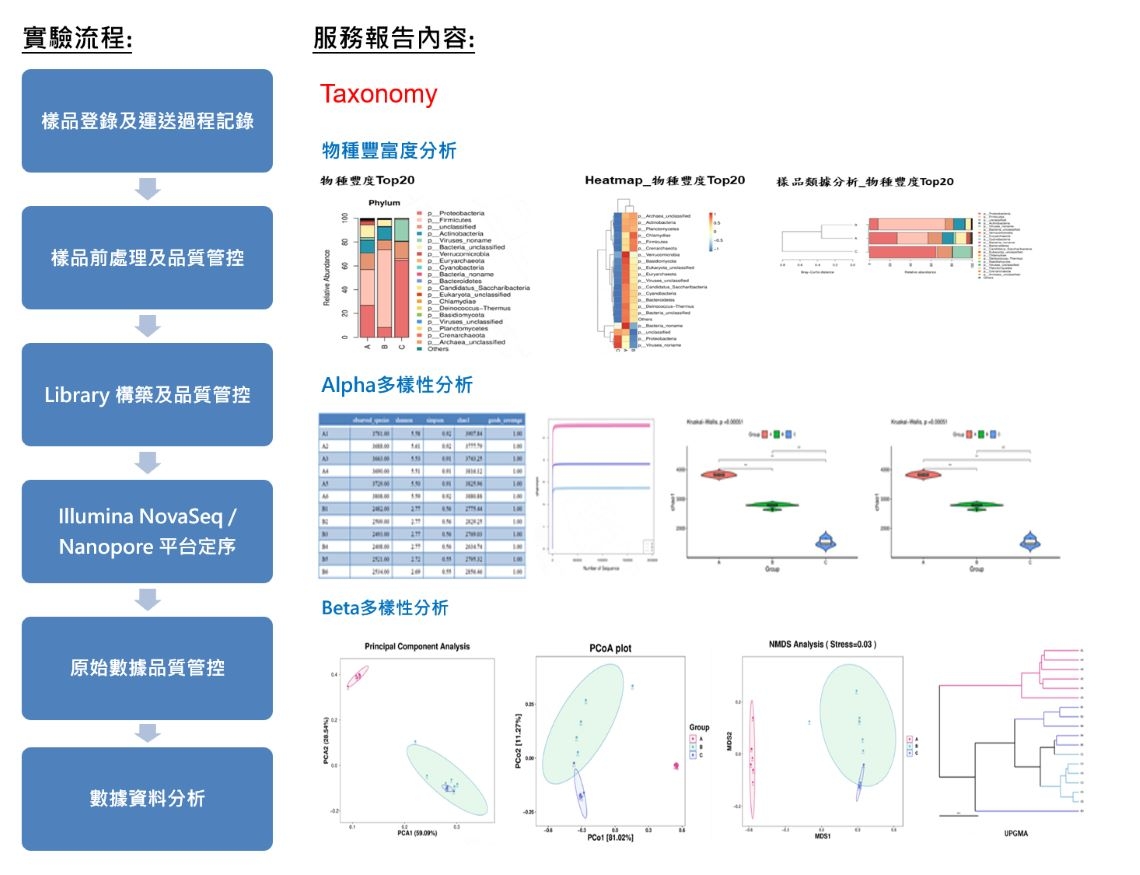
多源基因組學(宏基因組學) Whole Genome Metagenomics
多源基因體學(Metagenomics),又稱為環境基因組學(Environmental Genomics),是研究一整個微生物群體的科學,從環境樣品萃取其中微生物的DNA加以定序後,然後進行物種鑑定、親緣、基因功能、表型關聯等分析比對,以了解環境所含微生物的多樣性。
Whole Genome Metagenomics 是一種更全面的研究方法,可以獲得特定環境中整個微生物群落所有微生物的基因組序列資訊。擺脫微生物分離培養的技術限制,直接萃取環境樣品DNA進行定序,透過進行高通量DNA序列分析,獲得每個微生物的基因組資訊總和,用於研究微生物的菌種種類、分佈比例、親緣性、基因功能及代謝途徑等。藉由提供更深入的基因訊息,允許研究者能更全面地了解微生物群落的生態和代謝功能。
樣品需求:
樣品純度:OD 260/280值應在1.7~1.9之間
樣品濃度:最低濃度不低於 30 ng/µl
樣品總量:總量建議5 µg,至少不低於1.5 µg
樣品溶劑:可溶解在ddH2O或TE buffer(pH 8.0)中

RAD-seq (ezRAD-seq)
簡化基因組學 ezRAD-seq
RAD-seq (Restriction-site Associated DNA Sequence)係指利用限制性內切酶對基因組進行酶切,大幅降低基因組的複雜度,對特定長度的酶切片段(RAD-tag )進行高通量測序,快速篩選出高準確性的變異標記(SNPs),進行群體進化、遺傳圖譜構建及QTL 定位等分析。而ezRAD-seq 則是使用測序文庫套組來降低實驗操作複雜性的RAD-seq 技術,使該技術能迅速普及於一般實驗室的操作,不需太多的實驗設備和步驟即可獲得大量的SNPs 數據,能最大程度地降低時間和實驗成本。再者,ezRAD-seq 基於SNPs 的分子標記技術性價比高,穩定性好,特別適合大量樣本的分析。目前已廣泛應用於非模式物種的遺傳多樣性研究、分子育種、系統演化、群體基因組學等領域。
樣品需求:
樣品純度:OD 260/280值應在1.7~1.9之間
樣品總量:建議每個樣品總量不少於 2 μg DNA
樣品溶劑:可溶解在ddH2O或TE buffer(pH 8.0)中
ChIP-seq
染色質免疫沉澱測序 ChIP-Seq
染色質免疫沉澱法(Chromatin immunoprecipitation,ChIP)是研究體內DNA與蛋白質相互作用的重要工具,可靈敏地檢測目標蛋白與特異DNA片段的結合情況,由於真核生物基因組DNA是以染色質的形式存在,在染色質環境下,DNA與蛋白質相互作用闡明真核生物基因表現的基本途徑,因此透過ChIP技術使研究者得以瞭解染色質及其相結合的調控因子間的相互作用,通常用於轉錄因子結合位點或組蛋白特異性修飾位點的研究。而染色質免疫沉澱測序(ChIP-Seq)是將ChIP結合次世代測序(Next Generation Sequence,NGS)的技術,將取得的DNA片段進行高通量測序,獲得數百萬條序列並將其精確定位到基因組上,以檢測與組蛋白、轉錄因子等相互作用的DNA區段的生物訊息。
樣品需求:
樣品純度:OD 260/280比值應在1.7~1.9之間;RNA、蛋白質應去除乾淨。
樣品濃度:最低濃度不低於5 ng/μl (若樣本要進行QC,濃度不低於20 ng/μl)。
樣品總量:每個樣品總量不少於10 ng (若樣本要進行QC,總量不低於100 ng)。
樣品溶劑:可溶解在ddH2O或TE buffer(pH 8.0)中

RRBS-seq
簡化甲基化測序 RRBS-Seq
簡化甲基化測序 Reduced Representation Bisulfite Sequencing(RRBS)是一種準確、高效、經濟的DNA甲基化研究方法,通過酶切富集啟動子及CpG島區域,並進行Bisulfite測序,同時實現DNA甲基化狀態檢測的高解析度和測序數據的高利用率。DNA甲基化研究一直是疾病研究的熱點,與基因表達、表型性狀息息相關。RRBS作為一種高性價比的甲基化研究方法,在大規模臨床樣本的研究中具有廣泛的應用前景。
樣品需求:
樣品純度:OD 260/280比值應在1.7~1.9之間;RNA、蛋白質應去除乾淨
樣品濃度:最低濃度不低於50 µg/μl
樣品總量:每個樣品總量至少1 µg(建議 3 µg)
樣品溶劑:可溶解在ddH2O或TE buffer(pH 8.0)中
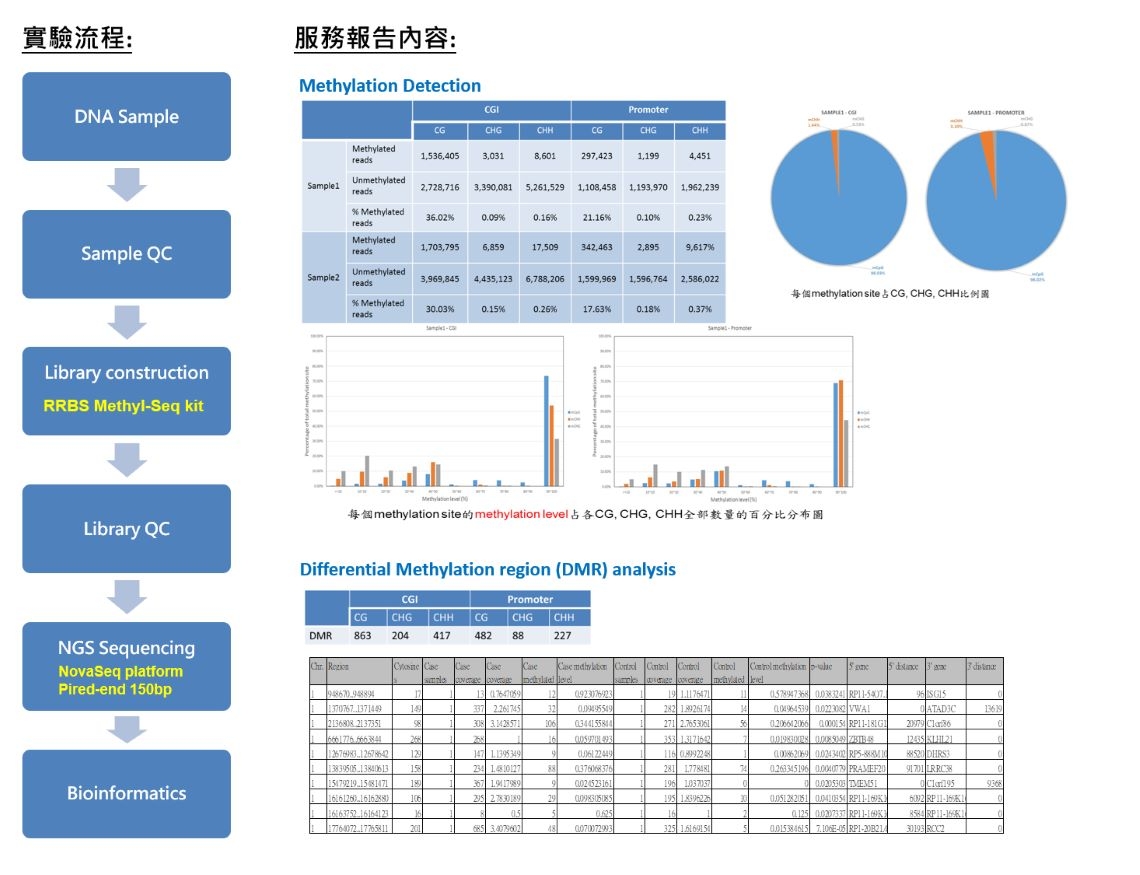
RNA-seq
轉錄組測序 RNA Sequencing
RNA測序(RNA sequencing),通常簡稱為RNA-seq,是分子生物學和基因體學中一種強大且廣泛應用的技術,用於分析細胞或組織的轉錄體(Transcriptome)。轉錄體是特定細胞或組織在特定時間內產生的RNA分子的完整集合,包括信使RNA(mRNA)、非編碼RNA和其他功能性RNA。
由於建庫與定序是一種「隨機」抽樣的過程,無法確保每一個片段都可以成功定序出來。再加上基因表現量各有不同,高表現的基因,會有大量的RNA片段,因此在定序過程中,有較高的機率被抽中;反之,低表現的基因,由於RNA片段量少,因此被抽中的機率相對低。隨著現在高通量的次世代基因定序(NGS)的發展,對於轉錄體的研究而言是個強而有力的工具。透過將長短不一的RNA片段打碎成150 bp的長度,再進行PCR反應的擴增,使得我們可以測得大量的數據,也能夠涵蓋完整的RNA資訊,以及獲得準確的量化資料。
RNA-seq可應用於諸如基因表達的分析、選擇性剪接分析、探索非編碼RNA(如 miRNA和 lncRNA等)。近期和 single cell技術結合發展出分析單個細胞的轉錄體,實現對組織內細胞異質性的研究。
樣本需求:
樣品純度:OD 260/280值應在1.9~2.1 之間;DNA、蛋白質應該去除乾淨
樣品總量:每個樣品總量建議1 µg以上(最少10 ng)
樣品溶劑:可溶解在ddH2O或TE buffer(pH 8.0)中,或是乾燥狀態下運送
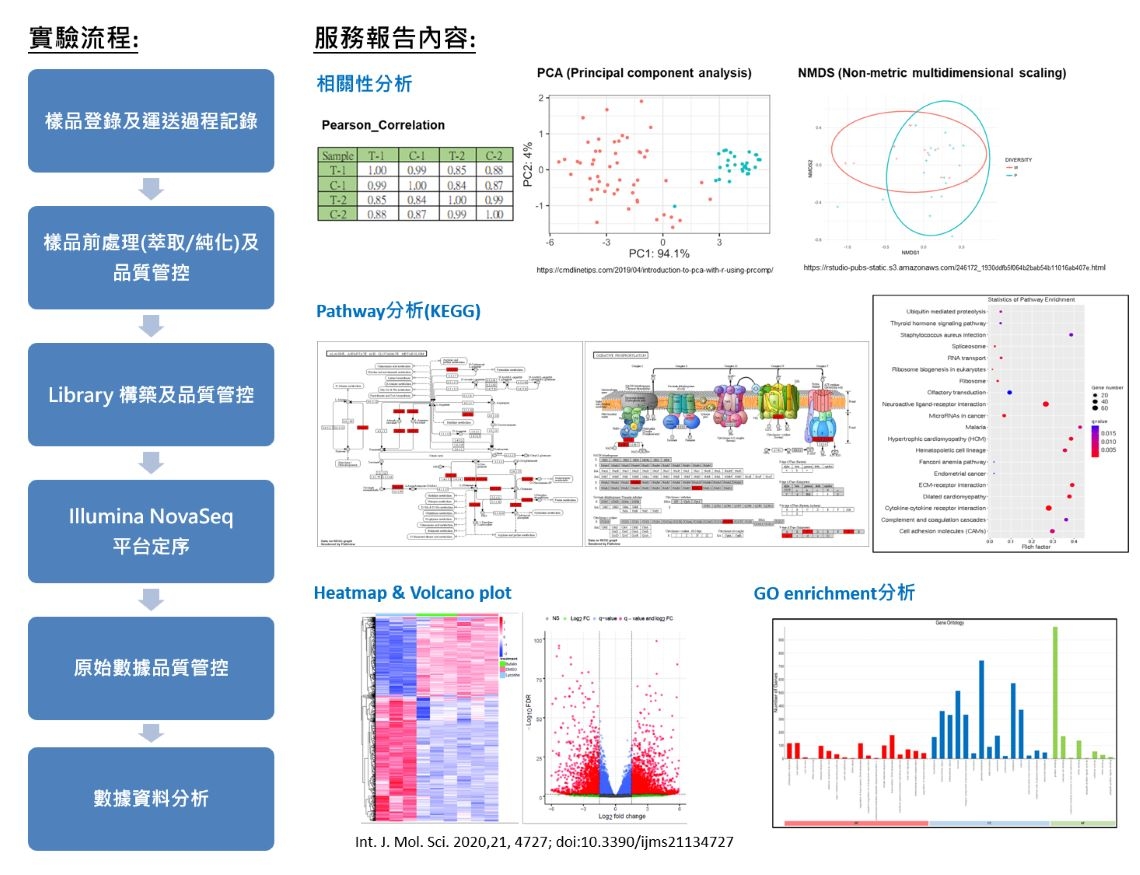
Small RNA seq
小片段RNA 測序 Small RNA Sequencing
microRNA(miRNA) 是一種內生的單股、非編碼的小片段 RNA,長度~22 個核苷酸,在生物體透過post-transcriptional 基因沈默的途徑,抑制蛋白質生合成,進而影響細胞發育、增生、分化、凋零等相關蛋白的表現。
自 1993 年首度發現 miRNA 至今,miRBase 註解已超過28645條miRNA,評估調控30%的人類基因,且每個miRNA又調控百個基因,故被認為與細胞分化、增殖與凋亡有關,因而影響幹細胞、免疫與癌症等系統,隨著癌症學的研究越多,發現在不同癌症中,miRNA表現狀況有所差異,其可調控腫瘤至發病過程中的基因,因此,可用以分類癌症、診斷癌症、瞭解預後結果,甚至發展治療方式。
樣本需求:
樣品純度:OD 260/280值應在1.8以上;DNA、蛋白質應該去除乾淨
樣品濃度:Total RNA最低濃度不低於25 ng/µl。(風險建庫可接受 2 ng/µl)
樣品總量:每個樣品總量不少於1000 ng 。(風險建庫可接受 10 ng)
樣品溶劑:可溶解在ddH2O或TE buffer(pH 8.0)中。或是乾燥狀態下運送

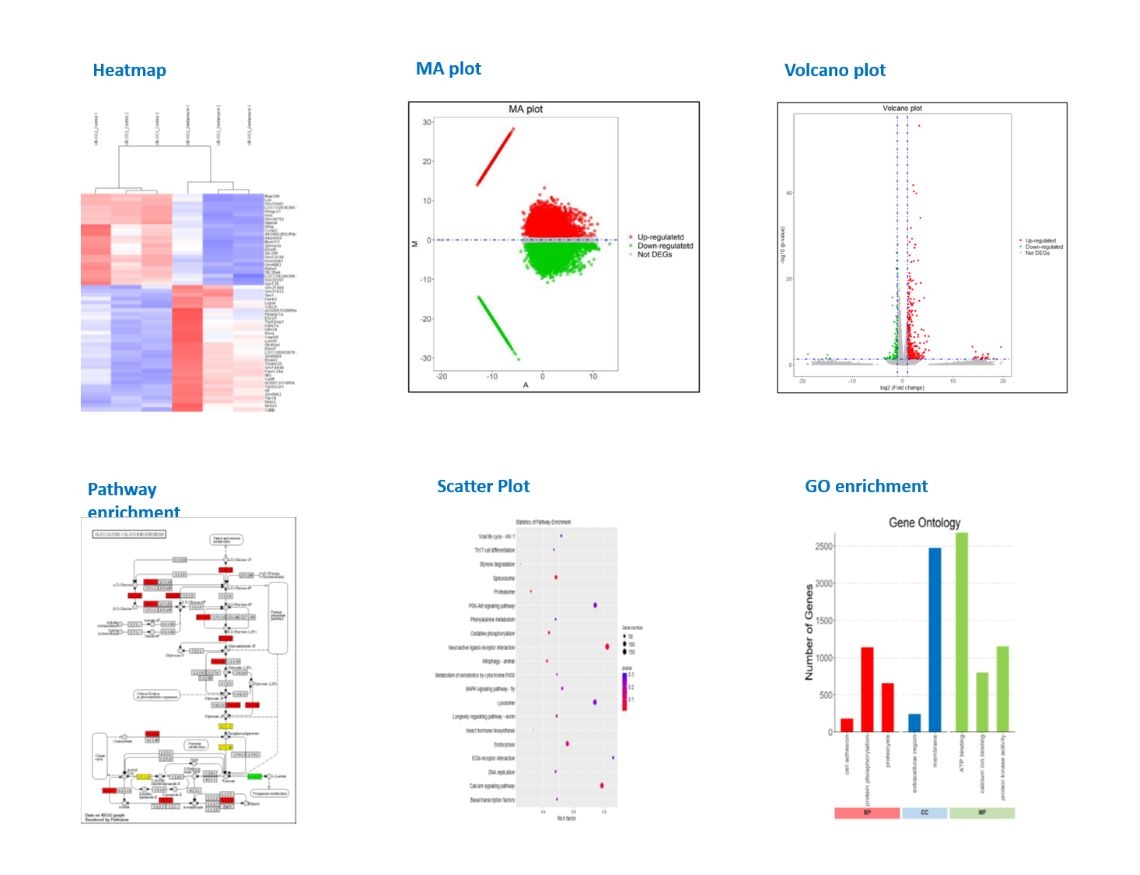
Ribo-seq
核醣體印跡測序 Ribo-seq
Ribo-seq(又稱Ribosome profiling)為以RNA-seq 作基礎所發展出的新技術,主要差別在Ribo-seq 於處理樣本的過程中,加入了RNase,將沒有核醣體保護的RNA 裂解,只分析有核醣體結合、當下正在進行轉譯的RNA片段(Ribosome-Protected RNA Fragments, RPFs),以此進一步了解轉譯調控(Translational Regulation)的機制。作為了解蛋白質合成調控的重要工具,我們的服務採用由IMMAGINA BioTechnology 研發的 RiboLace 技術,以磁珠針對轉譯中的核醣體作親和性純化,進而收集RPFs 進行高通量測序。相較於傳統Ribo-Seq,只需要少量的檢體(>0.3M cells),且不需切膠純化的步驟,將實驗時程由10 天簡化至2~3 天,除大大地降低檢體需求門檻,亦加快了數據取得時間。
樣品需求:
樣品純度:OD 260/280值應在1.7~1.9之間
樣品總量:建議每個樣品總量不少於 1.5 μg RNA
樣品濃度:濃度建議25 ng/μl 以上(濃度25 ng/μl 以上方能進行Bioanalyzer 品質檢測)
樣品溶劑:可溶解在ddH2O或TE buffer(pH 8.0)中

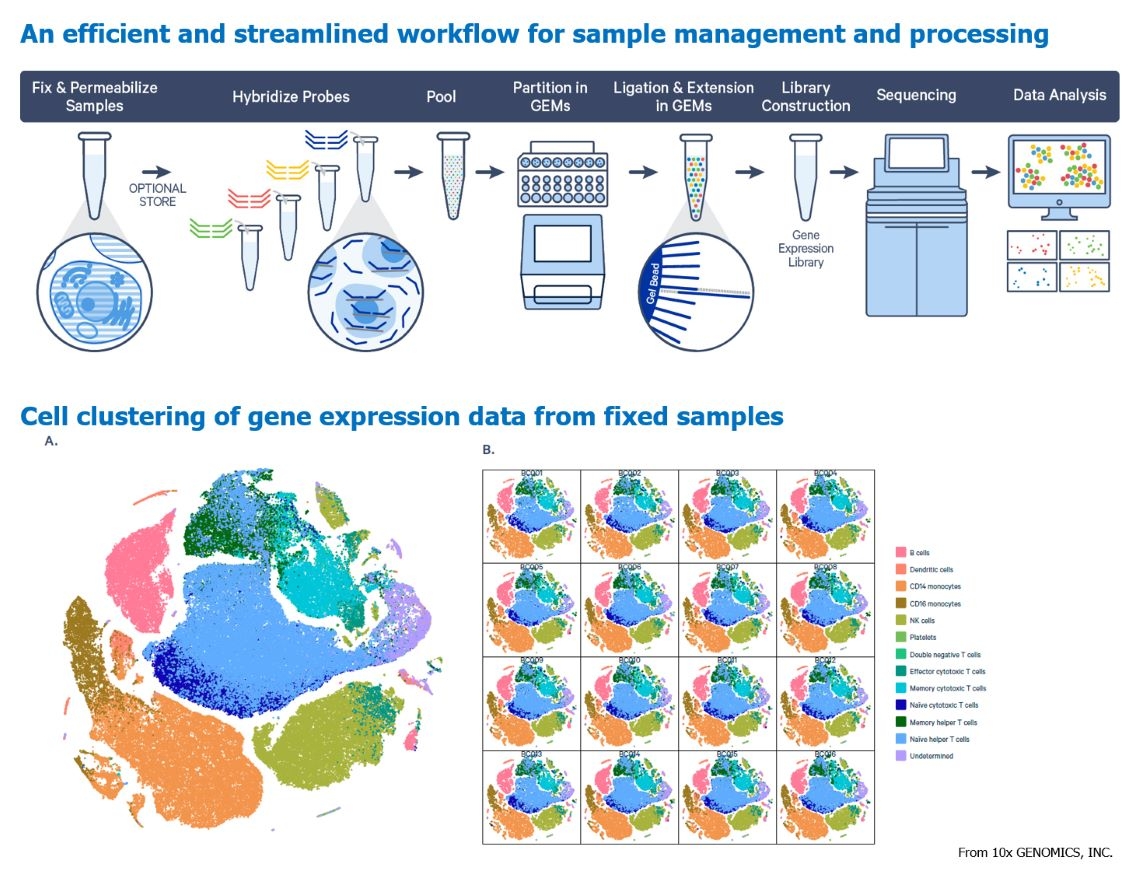
Single-cell RNA-seq
單細胞測序 Single-cell RNA-seq (scRNA-seq)
Single-cell RNA sequencing (scRNA-seq)單細胞RNA測序是一種新興且強大的分子生物學技術,用於分析單細胞層級的基因表現。傳統的RNA測序提供大量細胞中平均基因表現的資訊,但單細胞RNA測序提供每個單獨細胞的基因表現數據,使研究人員能夠檢查異質群體中單一細胞的基因表現譜,從而深入了解細胞多樣性、分化和其他生物過程,故此,單細胞RNA測序具有廣泛的應用領域,包括發育生物學、免疫學、神經科學、再生醫學和藥物發現。
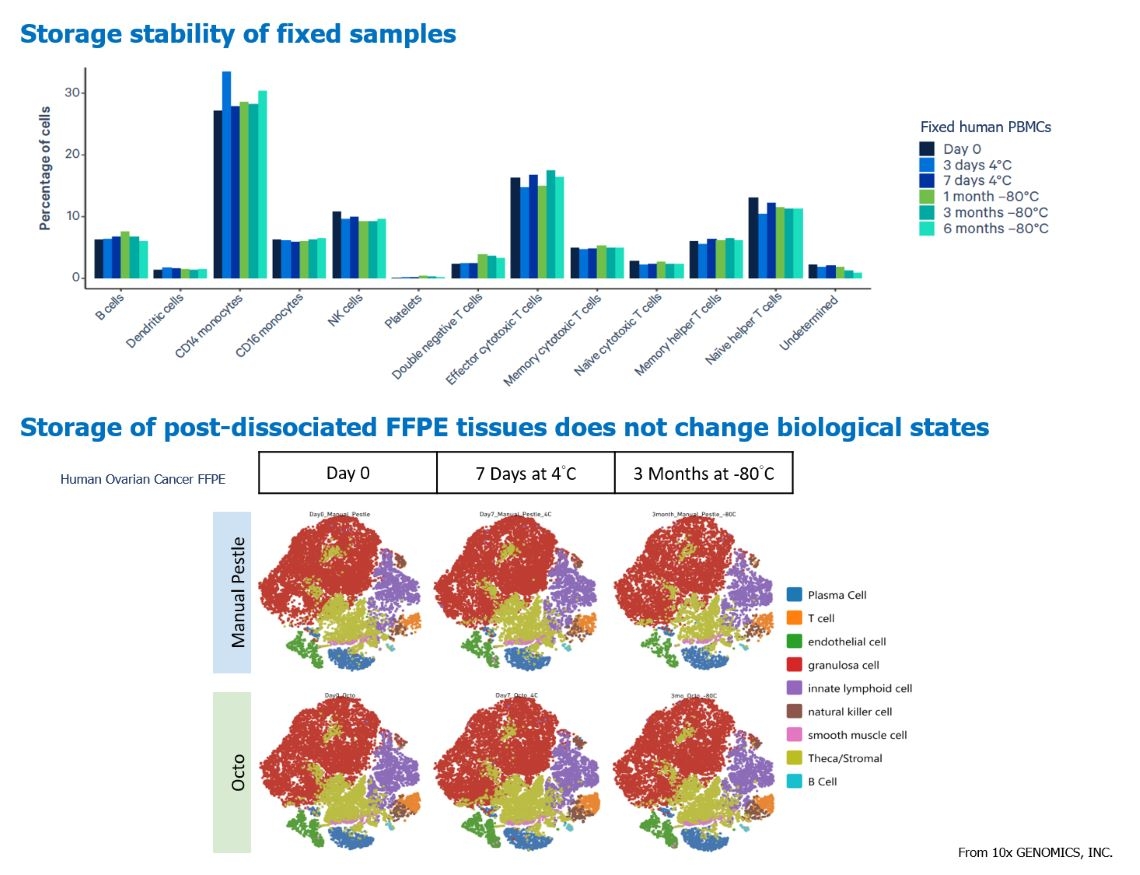
這一年來10x GENOMICS, INC更推出新的試劑套組,利用Fixation buffer 先固定活細胞,鎖住每顆細胞中RNA的表現狀態,後續再與偵測基因表現的探針組合反應,達到偵測每顆細胞基因表現量,被固定的細胞可以凍存於-80℃六個月仍可高度保持原有基因表達譜,甚至運用於FFPE檢體,故可降低以往僅能使用活細胞進行單細胞測序的反覆實驗動作、時間壓力,以及降低批次上機差異,並且減損及降低試劑耗損成本,將單細胞RNA測序的CP值最大化。
菌種鑑定服務
源資菌種鑑定檢測的優點:
1.基因鑑定方法分析細菌16S、真菌ITS,都可以鑑定到「種」的層次
2.單一菌株的菌盤,菌液均可做
3.收到樣本後,5個工作天出報告
4.專屬的數據管理系統提供客戶下載檔案